
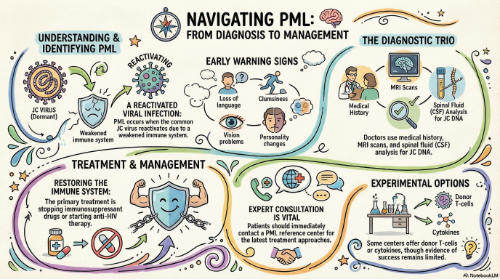
Progressive multifocal leukoencephalopathy (PML) is, in the general population, a rare but serious infection of the central nervous system (CNS). It results the reactivation of a common infection by a virus known as JC virus (abbreviated as JCV), and additional risk factors that include a weakened immune system and possibly genetic factors.
Prior infection with the JC virus is required for PML to develop. Antibodies against JCV are found in 50-70% of the general adult population, indicating prior infection, and in these people the virus will likely persist for life, but causing no obvious symptoms. Despite this high prevalence of JCV infection in the human population, the incidence of PML is very low.
The development of PML, while dependent on the presence of the JC virus, is the result of a failure from the immune system to keep JCV infection under control. This impairment of host immune function may occur in people with different conditions, including hematological tumors, such as lymphoma; therapeutic treatments that affect the immune system, such as immune suppressive therapies in transplanted people or in people with autoimmune disorders, or treatments with immunomodulant drugs aiming to control inflammation, such as in multiple sclerosis; HIV-infection; other conditions characterized by an abnormal immune control, such as idiopathic CD4 lymphocytopenia (ICL) or other congenital immune deficiencies. Indeed, the incidence of PML has increased in recent years due to the use of immunosuppressant or immunomodulant medications, such as natalizumab, used for treatment of multiple sclerosis, or rituximab, used to treat several tumors.
Although the mechanisms leading from a benign infection to such a severe disease as PML are not completely understood, there is evidence that, upon immune suppression, JCV escapes the host immune control and develops mutations in its genome that select for new viral variants with an advantage in terms of tropism and infection of brain cells. Thus, PML is eventually sustained by JCV genetic variants that are selected within an immunocompromised host. Finally, being PML such a rare disease, even in ‘at risk’ population, it is also possible that a genetic host predisposition plays a role in the chain of events leading to PML.
Initial symptoms of PML vary largely, depending on the site, number, and extension of the initial lesions, and are the result of a process of demyelination in the brain. These may include loss of language ability, changes in personality, memory loss, loss of coordination or clumsiness, vision problems, headaches, and seizures. Typically, the onset of symptoms is insidious, spanning up to several weeks. As the disease progresses, symptoms become worse, for example difficulties in hands movements may evolve into arms paresis, and severe disability or death will often result.
PML prognosis varies depending on several factors, including underlying comorbidities. If left unmanaged, the mortality rate is up to 50% within the first three months of diagnosis. In some cases, intervention can improve the chance of survival, although it is likely that some significant neurological deficits will be permanent. Since PML results from an impaired immune system, the only available treatment options are based on reconstitution of such impairment. This can be achieved, in people receiving immunosuppressive or immunomodulant drugs, by stopping their administration, or, in persons with HIV infection, through initiation of anti-HIV treatment. However, many patients may not respond to these approaches or not quickly enough to stop PML progression before development of severe deficits. Additional experimental immune-based approaches may also be used in some circumstances, but currently none of these has been shown to improve substantially PML outcome.